Intermolecular Dimerization Energies: MP2/6-311G** Calculations of the Dimerization of Chlorobenzene.
The Fock-ing Computational Chemists; Graeme Day and Mike Lewis
Introduction
The energy gained from the dimerization of two chlorobenzene molecules is crucial to undestanding the x-ray crystalographic structures of 4-methoxy-4'-chloroacetophenoneazine (MeO-Cl azine) (Figure 1). The inte rest in this azine stems from the alignment of the dipole moments in its crystral structure [1] (Figure 2), which makes it a nonlinear optically active material with various possible applications in photonics technology [2,3,4]. Our group has had a long s tanding
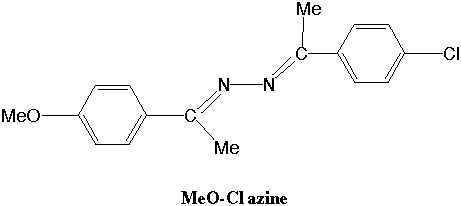
Figure 1
interest in nonlinear optical materials [5,6], and if we are to succeed in the synthesis of other nonlinear optical materials we first have to understand the crystal structure of MeO-Cl azine. A closer look at the crystal structure of MeO-Cl azine shows t hat each lattice consists of azine dimers (Figure 3), which constitute the smallest segments of the crystal that still depict the alignment of the dipole moments. It is our goal to understand the molecular forces that hold these dimers together and, there fore, this proposal will focus on dimerization energies.
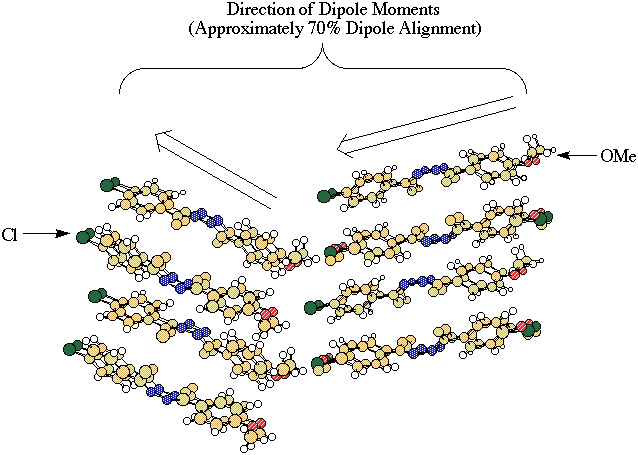
Figure 2
Goals & Objectives
A key step in understanding why MeO-Cl azine prefers to crystallize with its dipole moments aligned would be through theoretical study of the dimers shown in Figure 3, as well as the dimers where the dipo le moments are antiparallel aligned. The latter geometry could be simply obtained by interchanging the MeO- and Cl-substituents in one of the azines. This would tell us if it is more energetically favourable for the dipole moments to be parallel aligned o r antiparallel aligned in the dimer. If it turns out that the dipole moments prefer to be antiparallel aligned, as one would expect, then the observed crystal structure of MeO-Cl azine must be energetically favoured due to other molecular or crystal latti ce properties. Clearly, this is a problem that requires a quantum mechanical solution, as we are aware of no experimental techniques that would allow one to measure the energy holding a pair of molecules together in a crystal lattice.
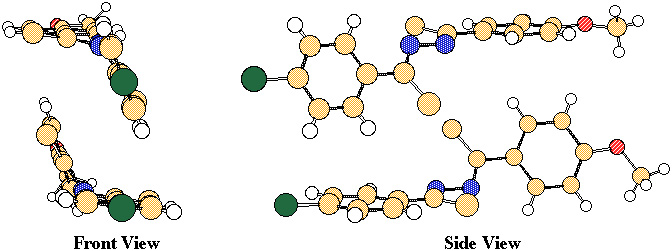
Figure 3
The size of the system presents an obvious obstacle to the computational approach to this problem. We propose to overcome this problem by studying only the interaction between the chloro-substituted phenyl rings. We arrive at chlorobenzene by substitution of the azine bridge para to the chlorine by a hydrogen atom. At first glance, this may appear to be a poor approach to the study of the azine dimerization because we usually envision donor-acceptor molecules as being fully conjugated. We would therefore expect the dipole moment associated with the chloro-substituted phenyl ring to be greatly affected by the amino- or methoxy-substituted phenyl rings. This, however, is not the case for MeO-Cl and similar azines b ecause they are not planar (Figure 3), and, thus, not conjugated. Moreover, it has recently been shown that the azine bridge in molecules such as MeO-Cl azine actually acts as a "conjugation stopper" [7], and therefore the electronic properties of the don or-substituted phenyl ring have little effect on the electronic properties of the acceptor-substituted phenyl ring. As a result, the energetics involved in the dimerization of the azines may be examined through the separate dimerizations of the donor and acceptor substituted phenyl rings.
Proposed Research
This study will focus on the intermolecular interactions of the chloro-substituted phenyl rings in MeO-Cl azine. Moreover, we will obtain a general result for the interaction of two chloro-substituted phen yl rings when the dipole moments are both parallel and antiparallel aligned. The first calculation that we will need to carry out is the geometry optimization of chlorobenzene. After this, the calculations that we will perform can be separated into three categories. First, we will optimize the dimerization of two chorobenzene molecules for perfect phenyl-phenyl T-contacts and perfect face-to-face contacts, in each case considering both the dipole parallel and dipole antiparallel geometries (Figure 4). The se calculations will only require the optimization of one parameter, i.e. the distance between the benzene rings, since all other parameters will be fixed to achieve perfect T-contacts and perfect face-to-face contacts. Second, we will optimize the geometries described in Figure 4 with no constraints on the angle or slippage between the phenyl rings. Third, we will perform single point calculations using the x-ray crystallographic geometries of the chloro-substituted phenyl rings in the MeO-Cl azin e. This will give us an estimate for the dimerization energy of the dipole parallel aligned chloro-substituted phenyl rings in MeO-Cl azine and provide some insight into the energetics involved in the crystal phase.
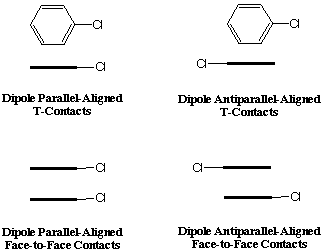
Figure 4
We intend to perform all of our calculations at the MP2(full)/6-311G** level of theory. We chose the MP2 method instead of the HF method because of the complex orbital interactions which we will be studying in the dimerization of polar molecules. Electron correlation is necessary to model these interactions adequately. Although perhaps more computationally efficient, the use of density functional theory is not considered in this study because of its inability to be systematically improved (as with MO meth ods) and because of its overemphasis on instantaneous electron-electron repulsion. Nor did we consider higher order perturbation or CI methods because these would be too computationally demanding for our needs.
The relatively large 6-311G** basis set was chosen because of the need for polarization functions in these polar molecules, with polarization functions added to hydrogens as well as heavy atoms because of the polarization which is undoubtedly necessary to describe the hydrogens in the field of the phenyl pi-electron cloud (i.e. in the edge-to-face dimers). A large, triply split valence basis set was also chosen to minimize basis set superposition errors (BSSE) in our dimerization energies. BSSE err ors would occur when the relative energies of the dimers are artificially lowered because a larger set of basis functions is made available to each of the molecules in the dimer, while not available to describe the electronic structure in the monomer calc ulations. These errors are large when small basis sets are used to describe the molecular orbitals, but vanish as the basis set is expanded. Cointerpoise correction calculations could be made by using ghost atoms from the geometry of the final dimer struc tures in the recalculation of monomer energies. We are aware of the overestimation of BSSE using counterpoise corrections, but they would give an error bound to our dimerization energies. However, these calculations are not included in our original propos al because such errors are expected to be small and the correction calculations would significantly increase (approximately double) our projected timeline.
The most important data to be obtained from this project are the relative energies of dimerization of the various dimer geometries optimized. The fact that the proposed project is mainly energetic in nature should not detract from its merits and importanc e in understanding the aggregation of this type of molecule in crystals. The comparison of dimerization energies should indicate the prefered orientation of such molecules and the strength of other intermolecular interactions in overcoming the unfavourabl e dipole parallel alignment observed in some crystal structures. Edge-to-face and face-to-face phenyl-phenyl interactions are known to be favourable in such systems and the resulting energies should provide insight into how molecules with the ability to interact in such ways will behave when they also have permanent dipoles.
The intermolecular distances obtained from the face-to-face and edge-to-face constrained geometry optimizations will be analyzed to observe how the dipole alignment affects the dimerization interactions. These calculations are proposed mainly for energeti c analysis. The more insightful geometrical parameters (i.e. intermolecular distances, phenyl-phenyl inter-ring angles, and lateral slippage of the rings) will be obtained from the unconstrained calculations and will show how the molecules concurre ntly optimize both the dipole-dipole (either attractive or repulsive) and the higher order intermolecular interactions. These geometrical parameters can then be compared to those found in crystal structures in order to estimate the importance of many-body interactions (both steric and electronic) in the crystal lattice.
We plan on using a Silicon Graphics Power Challenge-L supercomputer akin to the type we have here at the University of Missouri‚Columbia for these computations. A PC or a workstation would definitely be too small to complete this project in a reasonable l ength of time and, although a larger supercomputer, such as a Cray, would be a significantly faster, the Power Challenge-L will more than suffice for this project. The calculations that we described above involve one single point computation and nine geom etry optimizations. The single point calculation, with geometries from the crystal structures, should not take more than 8 hours. The optimization of chlorobenzene should take less than 12 hours, at a good guess, and the optimizations of the perfect T-con tacts and the perfect face-to-face contacts should also take less than 12 hours since we are only optimizing one parameter. The computationally demanding work will be the full optimizations of the phenyl-phenyl contact angles starting from edge-to-face an d face-to-face geometries. Even starting from the geometries of the perfect T-contacts and the perfect face-to-face contacts, we estimate that these computations could take two or more days. Therefore, we feel that a reasonable, and liberal, timeline for the completion of this project is between three and four weeks.